Hello Nextflow
Last updated on 2025-07-12 | Edit this page
Overview
Questions
- What is Nextflow?
- Why should I use a workflow management system?
- What are the features of Nextflow?
- What are the main components of a Nextflow script?
- How do I run a Nextflow script?
Objectives
- Understand a workflow management system.
- Understand the benefits of using a workflow management system.
- Explain the components of a Nextflow script.
- Run a Nextflow script.
Workflows
Analysing data involves a sequence of tasks, including gathering, cleaning, and processing data. This is what we refer to as data wrangling. This is an important part of research and something that should be documented. The sequence of tasks that picks up from the point that the data is ready to work with is a workflow or a pipeline. These workflows typically require using multiple software packages, sometimes running on different computing environments, such as desktop or a compute cluster. However, as workflows become larger and more complex, the management of the programming logic and software becomes difficult. Workflow Management Systems have been developed specifically to manage computational data-analysis workflows.
Workflow management systems
Reproducibility: Nextflow supports several container technologies, such as Docker and Singularity, as well as the package manager Conda. This, along with the integration of the GitHub code sharing platform, allows you to write self-contained pipelines, manage versions and to reproduce any previous result when re-run, including on different computing platforms.
Portability & interoperability: Nextflow’s syntax separates the functional logic (the steps of the workflow) from the configuration (how the scripts run). This allows the pipeline to be run on multiple platforms, e.g. local compute vs. a university compute cluster or a cloud service like AWS, without changing the steps of the workflow.
Simple parallelism: Nextflow is based on the dataflow programming model which greatly simplifies the splitting of tasks that can be run at the same time (parallelisation).
Continuous checkpoints & re-entrancy: All the intermediate results produced during the pipeline execution are automatically tracked. This allows you to resume its execution from the last successfully implemented step, no matter what the reason was for it stopping.
Processes, channels, and workflows
Nextflow workflows have three main parts: processes, channels, and workflows.
Processes describe a task to be run. A process script can be written in any scripting language that can be implemented by the Linux platform (Bash, Perl, Ruby, Python, R, etc.). Processes spawn a task for each complete input set. Each task is implemented independently and cannot interact with other tasks. The only way data can be passed between process tasks is via asynchronous queues, called channels.
Processes define inputs and outputs for a task. Channels are then used to manipulate the flow of data from one process to the next.
The interaction between processes, and ultimately the pipeline execution flow itself, is then explicitly defined in a workflow section.
Independent instances (tasks) of a process are run in parallel. Each task generates an output, which is passed to another channel and used as input for the next process. We’ll figure out how this works with some of the practice examples in the following sections.
Workflow implementation
While a process defines what command or script has to be
implemented, the runtime profile determines how that script
is actually run in the target system. If not otherwise specified,
processes are implemented on the local computer. The local runtime
profile is very useful for pipeline development, testing, and
small-scale workflows, but for large-scale computational pipelines, a
High Performance Cluster (HPC) or Cloud platform is often required.
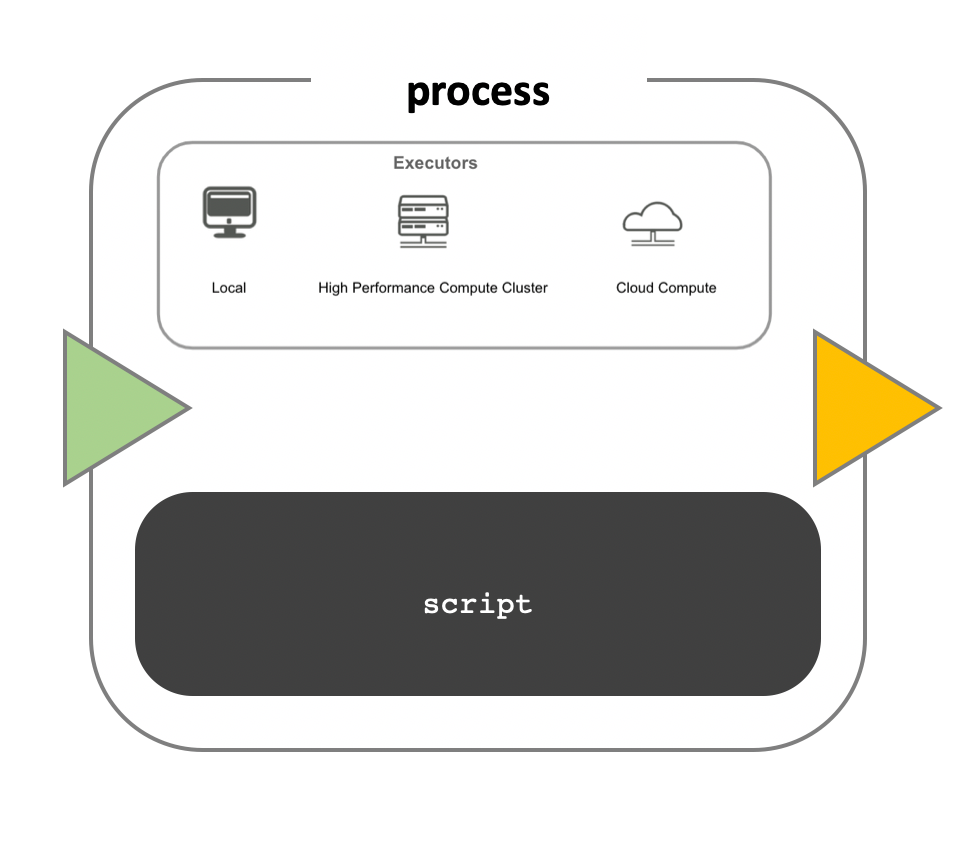
Runtime
Nextflow provides a separation between the pipeline’s functional logic and the underlying execution platform. This makes it possible to write a pipeline once, and then run it on your computer, compute cluster, or the cloud, without modifying the workflow, by defining the target platform in a configuration file. Note multiple configurations can be defined, and the choice is left to the user, a full list can be found here.
Your first script
We are now going to look at a sample Nextflow script that extracts a
folder with data files. First step involves navigating to the relevant
script, 02_hello_nextflow.nf in the current directory. The
instructions to set up the code material can be found under setup.
Task 2.1
The nextflow scripts for each lesson episode are available in the
scripts directory created during the course setup. Open the
02_hello_nextflow.nf script and consider each section of
the script.
Hint: Check that your terminal displays the following file path:
Use the cd command to change directory followed by the
folder name or relative path you want to navigate to, ex.
cd sgsss-workflow. If you want to go back up a level or
folder in your directory, you can type cd ..
This is a Nextflow script, which contains the following:
- An optional interpreter directive (“Shebang”) line, specifying the location of the Nextflow interpreter.
- A multi-line Nextflow comment, written using C style block comments, there are more comments later in the file.
- A pipeline parameter
params.inputwhich is given a default value, of the relative path to the location of a compressed archive of data, as a string. - A Nextflow channel
input_chused to read in data to the workflow. - An unnamed
workflowexecution block, which is the default workflow to run. - A call to the process
GENERATE_READS. - An operation on the process output, using the channel operator
.view(). - A Nextflow process block named
GENERATE_READS, which defines what the process does. - An
inputdefinition block that assigns theinputto the variableread, and declares that it should be interpreted as a file path. - An
outputdefinition block that uses the Linux/Unix standard output stream with file path from the script block. - A script block that contains the bash commands
printf '${targz}\\t'andtar -xzf $targz.
Running Nextflow scripts
To run a Nextflow script use the command
nextflow run <script_name>.
You should see output similar to the text shown below:
OUTPUT
N E X T F L O W ~ version 24.10.4
Launching `02_hello_nextflow.nf` [frozen_booth] DSL2 - revision: 8a3d1bb9c7
executor > local (1)
[52/af3b5c] GENERATE_READS (1) [100%] 1 of 1 ✔
[/workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school123_period1.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school123_period2.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school124_period1.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school124_period2.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school125_period1.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school125_period2.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school126_period1.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school126_period2.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school127_period1.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school127_period2.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school128_period1.RDS, /workspaces/training/work/52/af3b5ca0b401d80915bf823d321a7f/school128_period2.RDS]- The first line shows the Nextflow version number.
- The second line shows the run name
frozen_booth(adjective and scientist name) and revision id8a3d1bb9c7. - The third line tells you the process has been implemented locally
(
executor > local). - The next line shows the process id
52/af3b5c, process name, number of cpus, percentage task completion, and how many instances of the process have been run. Each instance is listed sequentially and separated by a,to demonstrate the flow of data as inputs. - The final line is the output of the
.view()operator.
Quick recap
- A workflow is a sequence of tasks that process a set of data, and a workflow management system (WfMS) is a computational platform that provides an infrastructure for the set-up, execution and monitoring of workflows.
- Nextflow scripts comprise of channels for controlling inputs and outputs, and processes for defining workflow tasks.
- You run a Nextflow script using the
nextflow runcommand.
Specifying an output directory in the script
To specify an output directory for a script use the parameter
publishDir in the definition of a process.
Task 2.3
Let’s edit the 02_hello_nextflow.nf script and specify
the output directory, where we want Nextflow to store data files that
were the result of decompressing the tar archive
folder.
The files that are extracted from the archive are stored in the
folder work that tracks the different tasks that are
launched as part of the pipeline. If we want a local copy of files that
are decompressed we can add an output directory with the following
code:
Note: You should always add a sensible default value to the pipeline parameter.
GROOVY
// 02_hello_nextflow.nf
process GENERATE_READS {
input:
path targz
publishDir "$projectDir", mode: "copy", overwrite: true
output:
path "*"
script:
"""
tar -xzf $targz
# Print file name
printf '${targz}\\t'
"""
}Here we’ve added the output directory "$projectDir" in
the process GENERATE_READS.
This step, publishDir "$projectDir", will add a
directory to output the decompressed files. The set of options that can
be specified are listed in the Nextflow
documentation. To access the value inside the process definition we
use $parameter syntax e.g. $projectDir.
You can also try to tweak the parameter, and specify a sub-folder
called tmp that will store the output of the process
"$projectDir/tmp".
The new line of code should read:
publishDir "$projectDir/tmp", mode: "copy", overwrite: true
How would this change where the output is stored?
- A workflow is a sequence of tasks that process a set of data.
- A workflow management system (WfMS) is a computational platform that provides an infrastructure for the set-up, execution and monitoring of workflows.
- Nextflow scripts comprise of channels for controlling inputs and outputs, and processes for defining workflow tasks.
- You run a Nextflow script using the
nextflow runcommand.